1. 課題を見つける/仮説を立てる
2. 実験や観察をしてデータを集める
3. データを整理する
4. データを解析して仮説を検証する
- 仮説の検証にはエビデンスが必要
- でも生物の表現型には ばらつきがある
- 実験や観察で得られた差や効果は偶然の産物じゃないの?
- これに答えるのが統計解析
- 推論や予測などにも使うよ
※資料作成は岩嵜航さん(東北大学)にご協力いただきました。
(左右キーで進みます!)
 (三中信宏)
(三中信宏) 



割合データは質的変数を変形したもの 
統計モデリング(重回帰分析)



※帰無仮説と対立仮説は排他的


\(Z\)統計量:\(\frac{\bar{X} - \mu}{\sqrt{\sigma^2/n}}\)…平均値の差を標準誤差\({\sqrt{\sigma^2/n}}\)で標準化
\(t\)統計量:\(\frac{\bar{X} - \mu}{\sqrt{\hat{\sigma}^2/n}}\)…母分散\(\sigma^2\)の代わりに不偏分散\(\hat{\sigma}^2\)を使う
他にも\(F\)統計量やカイ二乗統計量など
不適切な統計量(=方法)を用いると誤った\(p\)値が得られる


library(palmerpenguins) #penguinsを読み込み library(tidyverse) #tidyverseを読み込み head(penguins) #penguinsの最初を見せて!
## # A tibble: 6 x 8 ## species island bill_length_mm bill_depth_mm flipper_length_… body_mass_g sex ## <fct> <fct> <dbl> <dbl> <int> <int> <fct> ## 1 Adelie Torge… 39.1 18.7 181 3750 male ## 2 Adelie Torge… 39.5 17.4 186 3800 fema… ## 3 Adelie Torge… 40.3 18 195 3250 fema… ## 4 Adelie Torge… NA NA NA NA <NA> ## 5 Adelie Torge… 36.7 19.3 193 3450 fema… ## 6 Adelie Torge… 39.3 20.6 190 3650 male ## # … with 1 more variable: year <int>
penguins_adelie = penguins %>% filter(species == "Adelie") head(penguins_adelie)
## # A tibble: 6 x 8 ## species island bill_length_mm bill_depth_mm flipper_length_… body_mass_g sex ## <fct> <fct> <dbl> <dbl> <int> <int> <fct> ## 1 Adelie Torge… 39.1 18.7 181 3750 male ## 2 Adelie Torge… 39.5 17.4 186 3800 fema… ## 3 Adelie Torge… 40.3 18 195 3250 fema… ## 4 Adelie Torge… NA NA NA NA <NA> ## 5 Adelie Torge… 36.7 19.3 193 3450 fema… ## 6 Adelie Torge… 39.3 20.6 190 3650 male ## # … with 1 more variable: year <int>
gp = ggplot(data = penguins_adelie) +
geom_point(aes(x = sex, y = body_mass_g),
position = position_jitter(width = 0.05))
gp
## Warning: Removed 1 rows containing missing values (geom_point).

penguins_adelie = penguins %>% filter(species == "Adelie", !is.na(sex)) #NAを除く head(penguins_adelie)
## # A tibble: 6 x 8 ## species island bill_length_mm bill_depth_mm flipper_length_… body_mass_g sex ## <fct> <fct> <dbl> <dbl> <int> <int> <fct> ## 1 Adelie Torge… 39.1 18.7 181 3750 male ## 2 Adelie Torge… 39.5 17.4 186 3800 fema… ## 3 Adelie Torge… 40.3 18 195 3250 fema… ## 4 Adelie Torge… 36.7 19.3 193 3450 fema… ## 5 Adelie Torge… 39.3 20.6 190 3650 male ## 6 Adelie Torge… 38.9 17.8 181 3625 fema… ## # … with 1 more variable: year <int>
gp = ggplot(data = penguins_adelie) +
geom_point(aes(x = sex, y = body_mass_g),
position = position_jitter(width = 0.05))
gp



gp = ggplot(data = penguins_adelie) +
stat_qq(aes(sample = body_mass_g, color = sex)) +
facet_wrap( ~ sex, scales = "free")
gp

sw_test_results = penguins_adelie %>% #penguins_adelieを使う
group_by(sex) %>% #sexでグループ
summarise(statistic = shapiro.test(body_mass_g)$statistic,
#body_mass_gにshapiro.testを行い、そのstatistic値をstatisticという列に
p.value = shapiro.test(body_mass_g)$p.value, #同様
.groups = "drop") #グループを解除
head(sw_test_results)
## # A tibble: 2 x 3 ## sex statistic p.value ## <fct> <dbl> <dbl> ## 1 female 0.977 0.199 ## 2 male 0.983 0.416
var.test(data = penguins_adelie, body_mass_g ~ sex)
## ## F test to compare two variances ## ## data: body_mass_g by sex ## F = 0.60331, num df = 72, denom df = 72, p-value = 0.03356 ## alternative hypothesis: true ratio of variances is not equal to 1 ## 95 percent confidence interval: ## 0.3787186 0.9611059 ## sample estimates: ## ratio of variances ## 0.6033147
t.test(data = penguins_adelie, body_mass_g ~ sex)
## ## Welch Two Sample t-test ## ## data: body_mass_g by sex ## t = -13.126, df = 135.69, p-value < 2.2e-16 ## alternative hypothesis: true difference in means between group female and group male is not equal to 0 ## 95 percent confidence interval: ## -776.3012 -573.0139 ## sample estimates: ## mean in group female mean in group male ## 3368.836 4043.493
gp = ggplot(data = penguins_adelie) +
geom_point(aes(x = sex, y = body_mass_g), size = 0.8,
position = position_jitter(width = 0.07), alpha = 0.5) +
annotate("text", x = 1.5, y = 5200, label = "***", size = 5, color = "gray30") +
annotate("segment", x = 1.2, xend = 1.8, y = 5000, yend = 5000, color = "gray30") +
coord_cartesian(ylim = c(0, 6000))
gp
 ようやくグラフが完成!
ようやくグラフが完成!
#install.packages("easystats", repos = "https://easystats.r-universe.dev")
library("easystats")
report(t.test(data = penguins_adelie, body_mass_g ~ sex))
## Effect sizes were labelled following Cohen's (1988) recommendations. ## ## The Welch Two Sample t-test testing the difference of body_mass_g by sex (mean in group female = 3368.84, mean in group male = 4043.49) suggests that the effect is positive, statistically significant, and large (difference = 674.66, 95% CI [-776.30, -573.01], t(135.69) = -13.13, p < .001; Cohen's d = -2.25, 95% CI [-2.68, -1.82])
report(t.test(data = penguins_adelie, body_mass_g ~ sex)) %>% as.data.frame %>% #データフレームの形にして write_csv(file = "result_ttest.csv") %>% #csvに書き出し print() #表示
## Parameter | Group | Mean_Group1 | Mean_Group2 | Difference | 95% CI | t(135.69) | p | Method | Alternative | d | d CI ## ----------------------------------------------------------------------------------------------------------------------------------------------------------------------- ## body_mass_g | sex | 3368.84 | 4043.49 | 674.66 | [-776.30, -573.01] | -13.13 | < .001 | Welch Two Sample t-test | two.sided | -2.25 | [-2.68, -1.82]

t.test(data = penguins_adelie, body_mass_g ~ sex,
paired = FALSE, var.equal = TRUE) # paired = FALSEは省略可能
## ## Two Sample t-test ## ## data: body_mass_g by sex ## t = -13.126, df = 144, p-value < 2.2e-16 ## alternative hypothesis: true difference in means between group female and group male is not equal to 0 ## 95 percent confidence interval: ## -776.2484 -573.0666 ## sample estimates: ## mean in group female mean in group male ## 3368.836 4043.493
t.test(data = penguins_adelie, body_mass_g ~ sex,
paired = TRUE, var.equal = FALSE) # var.equal = FALSEは省略可能
## ## Paired t-test ## ## data: body_mass_g by sex ## t = -14.264, df = 72, p-value < 2.2e-16 ## alternative hypothesis: true difference in means is not equal to 0 ## 95 percent confidence interval: ## -768.944 -580.371 ## sample estimates: ## mean of the differences ## -674.6575
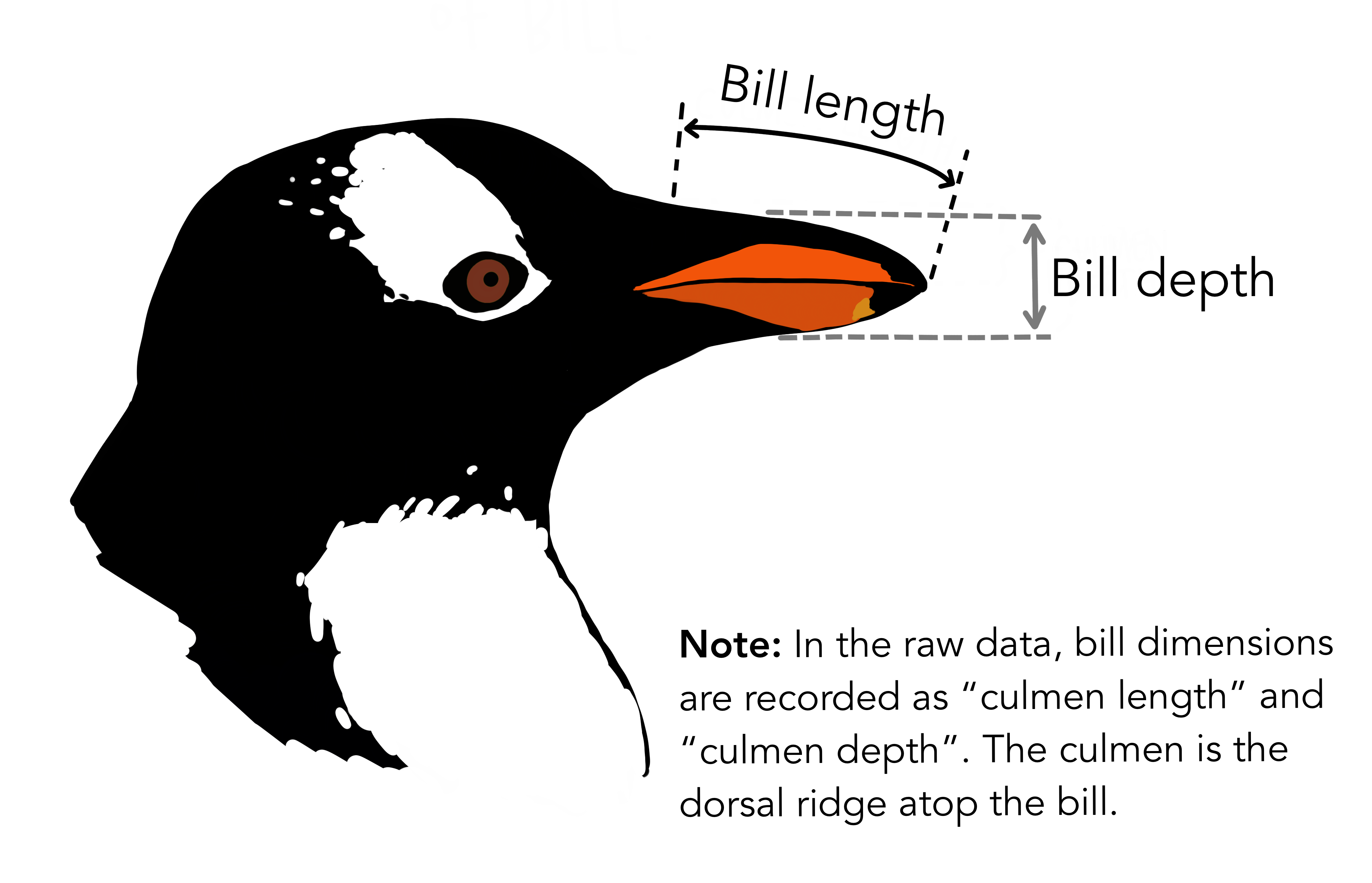
penguins_adelie = penguins %>% filter(species == "Adelie", !is.na(sex)) %>% #NAを除く mutate(ratio = bill_depth_mm/bill_length_mm) head(penguins_adelie)
## # A tibble: 6 x 9 ## species island bill_length_mm bill_depth_mm flipper_length_… body_mass_g sex ## <fct> <fct> <dbl> <dbl> <int> <int> <fct> ## 1 Adelie Torge… 39.1 18.7 181 3750 male ## 2 Adelie Torge… 39.5 17.4 186 3800 fema… ## 3 Adelie Torge… 40.3 18 195 3250 fema… ## 4 Adelie Torge… 36.7 19.3 193 3450 fema… ## 5 Adelie Torge… 39.3 20.6 190 3650 male ## 6 Adelie Torge… 38.9 17.8 181 3625 fema… ## # … with 2 more variables: year <int>, ratio <dbl>
gp = ggplot(data = penguins_adelie) +
geom_point(aes(x = sex, y = ratio),
position = position_jitter(width = 0.05))
gp

gp = ggplot(data = penguins_adelie) +
stat_qq(aes(sample = ratio, color = sex)) +
facet_wrap( ~ sex, scales = "free")
gp

sw_test_results = penguins_adelie %>% #penguins_adelieを使う
group_by(sex) %>% #sexでグループ
summarise(statistic = shapiro.test(ratio)$statistic,
p.value = shapiro.test(ratio)$p.value,
.groups = "drop")
head(sw_test_results)
## # A tibble: 2 x 3 ## sex statistic p.value ## <fct> <dbl> <dbl> ## 1 female 0.975 0.164 ## 2 male 0.954 0.00962
var.test(data = penguins_adelie, ratio ~ sex)
## ## F test to compare two variances ## ## data: ratio by sex ## F = 0.73443, num df = 72, denom df = 72, p-value = 0.1928 ## alternative hypothesis: true ratio of variances is not equal to 1 ## 95 percent confidence interval: ## 0.4610261 1.1699847 ## sample estimates: ## ratio of variances ## 0.7344341
まずインストールする(1行目の#を消して走らせる)
#install.packages("coin")
library(coin) # 読み込み
wilcox_test(data = penguins_adelie, ratio ~ sex, distribution = "exact")
## ## Exact Wilcoxon-Mann-Whitney Test ## ## data: ratio by sex (female, male) ## Z = 0.29941, p-value = 0.7661 ## alternative hypothesis: true mu is not equal to 0
まずインストールする(1行目の#を消して走らせる)
#install.packages("brunnermunzel")
library(brunnermunzel) # 読み込み
brunnermunzel.test(data = penguins_adelie, ratio ~ sex)
## ## Brunner-Munzel Test ## ## data: ratio by sex ## Brunner-Munzel Test Statistic = -0.29701, df = 143.58, p-value = 0.7669 ## 95 percent confidence interval: ## 0.3901081 0.5811811 ## sample estimates: ## P(X<Y)+.5*P(X=Y) ## 0.4856446
 阪大医学・老年・腎臓内科学HP
阪大医学・老年・腎臓内科学HPlibrary(palmerpenguins) #penguinsを読み込み library(tidyverse) #tidyverseを読み込み head(penguins) #penguinsの最初を見せて!
## # A tibble: 6 x 8 ## species island bill_length_mm bill_depth_mm flipper_length_… body_mass_g sex ## <fct> <fct> <dbl> <dbl> <int> <int> <fct> ## 1 Adelie Torge… 39.1 18.7 181 3750 male ## 2 Adelie Torge… 39.5 17.4 186 3800 fema… ## 3 Adelie Torge… 40.3 18 195 3250 fema… ## 4 Adelie Torge… NA NA NA NA <NA> ## 5 Adelie Torge… 36.7 19.3 193 3450 fema… ## 6 Adelie Torge… 39.3 20.6 190 3650 male ## # … with 1 more variable: year <int>
penguins_female = penguins %>% filter(sex == "female") head(penguins_female)
## # A tibble: 6 x 8 ## species island bill_length_mm bill_depth_mm flipper_length_… body_mass_g sex ## <fct> <fct> <dbl> <dbl> <int> <int> <fct> ## 1 Adelie Torge… 39.5 17.4 186 3800 fema… ## 2 Adelie Torge… 40.3 18 195 3250 fema… ## 3 Adelie Torge… 36.7 19.3 193 3450 fema… ## 4 Adelie Torge… 38.9 17.8 181 3625 fema… ## 5 Adelie Torge… 41.1 17.6 182 3200 fema… ## 6 Adelie Torge… 36.6 17.8 185 3700 fema… ## # … with 1 more variable: year <int>
gp = ggplot(data = penguins_female) +
geom_point(aes(x = species, y = body_mass_g),
position = position_jitter(width = 0.05))
gp

gp = ggplot(data = penguins_female) + stat_qq(aes(sample = body_mass_g, color = species)) + facet_wrap( ~ species, scales = "free") gp

sw_test_results = penguins_female %>% #penguins_adelieを使う
group_by(species) %>% #speciesでグループ
summarise(statistic = shapiro.test(body_mass_g)$statistic,
#body_mass_gにshapiro.testを行い、そのstatistic値をstatisticという列に
p.value = shapiro.test(body_mass_g)$p.value, #同様
.groups = "drop") #グループを解除
head(sw_test_results)
## # A tibble: 3 x 3 ## species statistic p.value ## <fct> <dbl> <dbl> ## 1 Adelie 0.977 0.199 ## 2 Chinstrap 0.963 0.306 ## 3 Gentoo 0.981 0.511
bartlett.test(data = penguins_female, body_mass_g ~ species)
## ## Bartlett test of homogeneity of variances ## ## data: body_mass_g by species ## Bartlett's K-squared = 0.19828, df = 2, p-value = 0.9056
summary(aov(data = penguins_female, body_mass_g ~ species))
## Df Sum Sq Mean Sq F value Pr(>F) ## species 2 60350016 30175008 393.2 <2e-16 *** ## Residuals 162 12430757 76733 ## --- ## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
TukeyHSD(aov(data = penguins_female, body_mass_g ~ species))
## Tukey multiple comparisons of means ## 95% family-wise confidence level ## ## Fit: aov(formula = body_mass_g ~ species, data = penguins_female) ## ## $species ## diff lwr upr p adj ## Chinstrap-Adelie 158.3703 22.32078 294.4197 0.0179471 ## Gentoo-Adelie 1310.9058 1195.64908 1426.1624 0.0000000 ## Gentoo-Chinstrap 1152.5355 1011.00620 1294.0648 0.0000000
gp = ggplot(data = penguins) +
geom_point(aes(x = species, y = body_mass_g), size = 0.5,
position = position_jitter(width = 0.05), alpha = 0.3) +
annotate("text", x = c(1, 2, 3), y = 7000, label = c("a", "b", "c")) +
coord_cartesian(ylim = c(0, 8000))
gp
## Warning: Removed 2 rows containing missing values (geom_point).
 できあがり!
できあがり!
kruskal.test(data = penguins_female, body_mass_g ~ species)
## ## Kruskal-Wallis rank sum test ## ## data: body_mass_g by species ## Kruskal-Wallis chi-squared = 115.24, df = 2, p-value < 2.2e-16
まずインストールする(1行目の#を消して走らせる)
#install.packages("NSM3")
library(NSM3)
pSDCFlig(penguins_female$body_mass_g, penguins_female$species, method = "Asymptotic")
## Ties are present, so p-values are based on conditional null distribution. ## Group sizes: 73 34 58 ## Using the Asymptotic method: ## ## For treatments Adelie - Chinstrap, the Dwass, Steel, Critchlow-Fligner W Statistic is 3.8595. ## The smallest experimentwise error rate leading to rejection is 0.0175 . ## ## For treatments Adelie - Gentoo, the Dwass, Steel, Critchlow-Fligner W Statistic is 13.877. ## The smallest experimentwise error rate leading to rejection is 0 . ## ## For treatments Chinstrap - Gentoo, the Dwass, Steel, Critchlow-Fligner W Statistic is 11.2606. ## The smallest experimentwise error rate leading to rejection is 0 . ##



Coded by Heavy Watal

library(palmerpenguins) #penguinsを読み込み library(tidyverse) #tidyverseを読み込み head(penguins) #penguinsの最初を見せて!
## # A tibble: 6 x 8 ## species island bill_length_mm bill_depth_mm flipper_length_… body_mass_g sex ## <fct> <fct> <dbl> <dbl> <int> <int> <fct> ## 1 Adelie Torge… 39.1 18.7 181 3750 male ## 2 Adelie Torge… 39.5 17.4 186 3800 fema… ## 3 Adelie Torge… 40.3 18 195 3250 fema… ## 4 Adelie Torge… NA NA NA NA <NA> ## 5 Adelie Torge… 36.7 19.3 193 3450 fema… ## 6 Adelie Torge… 39.3 20.6 190 3650 male ## # … with 1 more variable: year <int>
gp = ggplot(data = penguins_adelie) + geom_point(aes(x = body_mass_g, y = flipper_length_mm)) gp

glm_result = glm(flipper_length_mm ~ body_mass_g, #線形予測子
family = gaussian(link = "identity"), #誤差分布とリンク関数
data = penguins_adelie) #データ
summary(glm_result) #結果を格納したglm_resultの詳細を表示する
## ## Call: ## glm(formula = flipper_length_mm ~ body_mass_g, family = gaussian(link = "identity"), ## data = penguins_adelie) ## ## Deviance Residuals: ## Min 1Q Median 3Q Max ## -14.426 -3.669 0.239 3.422 17.955 ## ## Coefficients: ## Estimate Std. Error t value Pr(>|t|) ## (Intercept) 1.656e+02 3.918e+00 42.27 < 2e-16 *** ## body_mass_g 6.610e-03 1.049e-03 6.30 3.4e-09 *** ## --- ## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1 ## ## (Dispersion parameter for gaussian family taken to be 33.57457) ## ## Null deviance: 6167.5 on 145 degrees of freedom ## Residual deviance: 4834.7 on 144 degrees of freedom ## AIC: 931.33 ## ## Number of Fisher Scoring iterations: 2
gp = ggplot(data = penguins_adelie, aes(x = body_mass_g, y = flipper_length_mm)) +
geom_point(alpha = 0.5) + #こうするとaes()はggplot()の中の設定が適用される
geom_smooth(method = "glm", method.args = list(family = gaussian(link = "identity"))) +
annotate("text", x = 4200, y = 210,
label = "(Y ~ 0.006677X + 165.2, p = 3.4e-09)", size = 4)
gp

report(glm_result) %>% as.data.frame() %>% #データフレーム型にする write_csv(file = "result_glm.csv") %>% # CSVで出力 print() #ここでも表示
## Parameter | Coefficient | 95% CI | t(144) | p | Std. Coef. | Std. Coef. 95% CI | Fit ## -------------------------------------------------------------------------------------------------------- ## (Intercept) | 165.60 | [157.92, 173.28] | 42.27 | < .001 | -1.92e-15 | [-0.14, 0.14] | ## body mass g | 6.61e-03 | [ 0.00, 0.01] | 6.30 | < .001 | 0.46 | [ 0.32, 0.61] | ## | | | | | | | ## AIC | | | | | | | 931.33 ## BIC | | | | | | | 940.28 ## R2 | | | | | | | 0.22 ## Sigma | | | | | | | 5.79

shapiro.test(glm_result$residuals) #残差の正規性をShapiro検定
## ## Shapiro-Wilk normality test ## ## data: glm_result$residuals ## W = 0.99179, p-value = 0.562
Coded by Heavy Watal


Coded by Heavy Watal

Coded by Heavy Watal

\(Prob(X = k \mid \lambda) = \frac {\lambda^k e^{-\lambda}} {k!}\)
Coded by Heavy Watal
seed =
read_csv("https://kuboweb.github.io/-kubo/stat/iwanamibook/fig/poisson/data3a.csv") %>%
rename(no.seed = y, plant_size = x, treatment = f)
head(seed)
## # A tibble: 6 x 3 ## no.seed plant_size treatment ## <dbl> <dbl> <chr> ## 1 6 8.31 C ## 2 6 9.44 C ## 3 6 9.5 C ## 4 12 9.07 C ## 5 10 10.2 C ## 6 4 8.32 C
gp = ggplot(data = seed) + geom_point(aes(x =plant_size , y = no.seed)) gp

glm_result = glm(no.seed ~ plant_size,
family = poisson(link = "log"),
data = seed)
summary(glm_result)
## ## Call: ## glm(formula = no.seed ~ plant_size, family = poisson(link = "log"), ## data = seed) ## ## Deviance Residuals: ## Min 1Q Median 3Q Max ## -2.3679 -0.7348 -0.1775 0.6987 2.3760 ## ## Coefficients: ## Estimate Std. Error z value Pr(>|z|) ## (Intercept) 1.29172 0.36369 3.552 0.000383 *** ## plant_size 0.07566 0.03560 2.125 0.033580 * ## --- ## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1 ## ## (Dispersion parameter for poisson family taken to be 1) ## ## Null deviance: 89.507 on 99 degrees of freedom ## Residual deviance: 84.993 on 98 degrees of freedom ## AIC: 474.77 ## ## Number of Fisher Scoring iterations: 4
\(\beta_0\): 1.29172 (Pr(>|t|)=0.000383)
\(\beta_1\): 0.07566 (Pr(>|t|)=0.033580)
種子数への植物サイズの効果は有意である
\(Y = e^{(1.29172 + 0.07566 X)}\)に回帰できる
gp = ggplot(data = seed) +
geom_point(aes(x =plant_size , y = no.seed)) +
geom_smooth(aes(x =plant_size , y = no.seed),
method = glm,
method.args = list(family = poisson(link = "log")))
gp


Coded by Heavy Watal

\({Prob}(X = k \mid n,~p) = \binom n k p^k (1 - p)^{n - k}\)
Coded by Heavy Watal
seed2=
read_csv("https://kuboweb.github.io/-kubo/stat/iwanamibook/fig/binomial/data4a.csv") %>%
rename(observed_seed = N, survived_seed = y, plant_size = x, treatment = f)
head(seed2)
## # A tibble: 6 x 4 ## observed_seed survived_seed plant_size treatment ## <dbl> <dbl> <dbl> <chr> ## 1 8 1 9.76 C ## 2 8 6 10.5 C ## 3 8 5 10.8 C ## 4 8 6 10.9 C ## 5 8 1 9.37 C ## 6 8 1 8.81 C
gp = ggplot(data = seed2) + geom_point(aes(x = plant_size , y = survived_seed, color = treatment)) gp

seed3 = seed2 %>% mutate(dead_seed = observed_seed-survived_seed) head(seed3)
## # A tibble: 6 x 5 ## observed_seed survived_seed plant_size treatment dead_seed ## <dbl> <dbl> <dbl> <chr> <dbl> ## 1 8 1 9.76 C 7 ## 2 8 6 10.5 C 2 ## 3 8 5 10.8 C 3 ## 4 8 6 10.9 C 2 ## 5 8 1 9.37 C 7 ## 6 8 1 8.81 C 7
glm_result = glm(cbind(survived_seed, dead_seed) ~ plant_size,
family = binomial(link = "logit"),
data = seed3)
summary(glm_result)
## ## Call: ## glm(formula = cbind(survived_seed, dead_seed) ~ plant_size, family = binomial(link = "logit"), ## data = seed3) ## ## Deviance Residuals: ## Min 1Q Median 3Q Max ## -3.3998 -1.1536 0.2472 1.1787 3.1231 ## ## Coefficients: ## Estimate Std. Error z value Pr(>|z|) ## (Intercept) -13.7785 1.0773 -12.79 <2e-16 *** ## plant_size 1.4626 0.1107 13.21 <2e-16 *** ## --- ## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1 ## ## (Dispersion parameter for binomial family taken to be 1) ## ## Null deviance: 499.23 on 99 degrees of freedom ## Residual deviance: 217.17 on 98 degrees of freedom ## AIC: 364.35 ## ## Number of Fisher Scoring iterations: 5
glm_result = glm(cbind(survived_seed, dead_seed) ~ plant_size + treatment,
family = binomial(link = "logit"),
data = seed3)
summary(glm_result)
## ## Call: ## glm(formula = cbind(survived_seed, dead_seed) ~ plant_size + ## treatment, family = binomial(link = "logit"), data = seed3) ## ## Deviance Residuals: ## Min 1Q Median 3Q Max ## -3.2584 -0.7948 0.1753 0.8757 3.1589 ## ## Coefficients: ## Estimate Std. Error z value Pr(>|z|) ## (Intercept) -19.5361 1.4138 -13.82 <2e-16 *** ## plant_size 1.9524 0.1389 14.06 <2e-16 *** ## treatmentT 2.0215 0.2313 8.74 <2e-16 *** ## --- ## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1 ## ## (Dispersion parameter for binomial family taken to be 1) ## ## Null deviance: 499.23 on 99 degrees of freedom ## Residual deviance: 123.03 on 97 degrees of freedom ## AIC: 272.21 ## ## Number of Fisher Scoring iterations: 5
gp = ggplot(data = seed2) +
geom_point(aes(x = plant_size , y = survived_seed/observed_seed, color = treatment)) +
geom_smooth(aes(x = plant_size , y = survived_seed/observed_seed, color = treatment),
method = glm, method.args = list(family = binomial(link = "logit")))
gp

#install.packages("MASS") #まずインストール
library(MASS) #まず読み込み
head(birthwt, n = 2) #MASSに入っているbirthwtというサンプルデータを使う
## low age lwt race smoke ptl ht ui ftv bwt ## 85 0 19 182 2 0 0 0 1 0 2523 ## 86 0 33 155 3 0 0 0 0 3 2551
gp = ggplot(data = birthwt, aes(x = lwt , y = low)) + geom_point() gp

glm_result = glm(low ~ lwt,
family = binomial(link = "logit"),
data = birthwt)
summary(glm_result)
## ## Call: ## glm(formula = low ~ lwt, family = binomial(link = "logit"), data = birthwt) ## ## Deviance Residuals: ## Min 1Q Median 3Q Max ## -1.0951 -0.9022 -0.8018 1.3609 1.9821 ## ## Coefficients: ## Estimate Std. Error z value Pr(>|z|) ## (Intercept) 0.99831 0.78529 1.271 0.2036 ## lwt -0.01406 0.00617 -2.279 0.0227 * ## --- ## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1 ## ## (Dispersion parameter for binomial family taken to be 1) ## ## Null deviance: 234.67 on 188 degrees of freedom ## Residual deviance: 228.69 on 187 degrees of freedom ## AIC: 232.69 ## ## Number of Fisher Scoring iterations: 4
gp = ggplot(data = birthwt, aes(x = lwt , y = low)) +
geom_point() +
geom_smooth(method = glm,
method.args = list(family = binomial(link = "logit")))
gp

正規分布(gaussian)…確率変数の和、平均値
二項分布(binomial)…成功率p、試行回数nのうちの成功回数
ポアソン分布(poisson)…単位時間あたり平均λ回起こる事象の発生回数
ガンマ分布(Gamma)…ポアソン過程でk回起こるまでの待ち時間
他にinverse.gaussian, quasi, quasibinomial, quasipoisson.
それぞれの分布やリンク関数の形を調べて選ぼう
penguins_sexratio_sum = penguins %>%
group_by(species, sex) %>% # speciesとsexごとにグループ分け
summarise(length = length(species), # N数を数える
.groups = "drop") %>% # groupを解除
filter(!is.na(sex)) %>% # sex == NAを除去
print() # 表示
## # A tibble: 6 x 3 ## species sex length ## <fct> <fct> <int> ## 1 Adelie female 73 ## 2 Adelie male 73 ## 3 Chinstrap female 34 ## 4 Chinstrap male 34 ## 5 Gentoo female 58 ## 6 Gentoo male 61
gp = ggplot(data = penguins_sexratio_sum, aes(x = species, y = length)) + geom_bar(aes(fill = sex), stat = "identity", position = "fill") + scale_y_continuous(labels = scales::percent) + coord_flip() # X軸とY軸を逆にする gp

penguins_sexratio = penguins %>% filter(!is.na(sex)) # NAを除く chisq.test(penguins_sexratio$sex, penguins_sexratio$species)
## ## Pearson's Chi-squared test ## ## data: penguins_sexratio$sex and penguins_sexratio$species ## X-squared = 0.048607, df = 2, p-value = 0.976